

1 – Aspectos genéticos e embriológicos da Displasia Cleidocraniana
Mutação no gene RUNX2 (localizado no cromossomo 6p21); Tipo de herança: autossômica dominante; Papel do RUNX2 na diferenciação osteoblástica; Alterações no desenvolvimento embrionário dos ossos derivados do mesênquima membranoso (ex: crânio e clavículas); Anomalias de migração e ossificação intramembranosa.
A Displasia Cleidocraniana (DCC) é uma condição genética rara que compromete o desenvolvimento dos ossos que se originam predominantemente por ossificação intramembranosa, como o crânio, as clavículas e parte da face média. As bases da doença envolvem mutações moleculares que impactam diretamente o desenvolvimento embrionário do sistema esquelético, afetando tanto a diferenciação celular quanto a organização tecidual.
A causa principal da DCC é uma mutação heterozigótica no gene RUNX2, localizado no cromossomo 6p21, que codifica um fator de transcrição essencial para a diferenciação osteoblástica. Esse fator regula a transcrição de genes estruturais e reguladores da matriz óssea, como colágeno tipo I, osteocalcina, fosfatase alcalina, entre outros.
A condição apresenta padrão de herança autossômica dominante, ou seja, uma única cópia mutada do gene é suficiente para causar a síndrome. Apesar disso, a expressividade clínica é variável, podendo haver grande diversidade de sinais clínicos entre os indivíduos afetados, inclusive dentro da mesma família.
O RUNX2 atua como um fator mestre da osteogênese, sendo o principal responsável pela ativação da cascata genética que converte células-tronco mesenquimais em osteoblastos funcionais. Esse processo é fundamental para a formação óssea adequada, principalmente nos ossos que se desenvolvem por ossificação intramembranosa, onde não há fase cartilaginosa intermediária.
Quando o RUNX2 é funcionalmente deficiente, a diferenciação osteoblástica é interrompida, e os precursores mesenquimais não conseguem amadurecer em células formadoras de osso, resultando em formação incompleta ou ausente da matriz óssea.
Durante o desenvolvimento embrionário, ossos como as clavículas, a calota craniana e partes da face se originam a partir de mesênquima membranoso, que normalmente deveria se transformar diretamente em osso por ossificação intramembranosa.
Na DCC, não há falha primária na migração das células mesenquimais ou das células da crista neural — essas células ainda conseguem atingir seus destinos anatômicos. No entanto, a deficiência do RUNX2 compromete a arquitetura do microambiente osteogênico, afetando indiretamente a organização espacial, polarização e funcionalidade celular. Como resultado, a mineralização óssea é deficiente, e a formação esquelética se torna incompleta ou retardada.
Esse desfecho é particularmente evidente nos ossos membranosos, cuja formação depende quase exclusivamente da diferenciação mediada por RUNX2, em contraste com ossos endocondrais, que podem permanecer relativamente preservados.
Principais manifestações ósseas:
- Clavículas ausentes ou hipoplásica: Permitem mobilidade excessiva dos ombros, com possibilidade de aproximação medial anormal.
- Fontanelas amplas e fechamento tardio das suturas cranianas: Devido à ossificação deficiente da calota craniana, essas aberturas permanecem patentes por anos.
- Presença de ossos wormianos: Pequenos ossículos suturais acessórios visíveis entre as placas cranianas.
- Hipoplasia da face média com projeção frontal proeminente: A formação incompleta da maxila e dos ossos zigomáticos acentua a proeminência frontal.
- Alterações dentárias importantes: Dentes supranumerários, atraso na erupção dentária e falhas de formação radicular são achados frequentes.
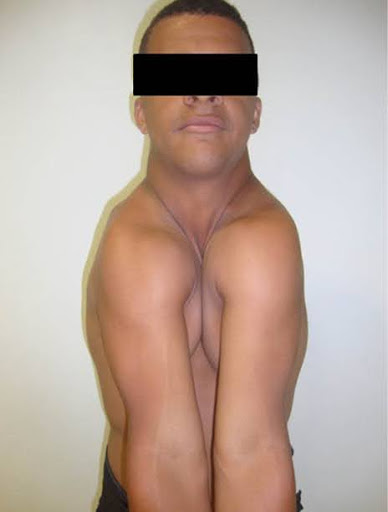
Imagem 1: Ausência de clavículas
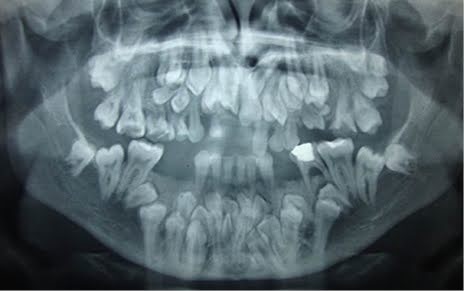
Imagem 2: Presença de dentes supranumerários
2 – Histologia e fisiologia óssea afetadas pela DCC
Tipos de ossificação: intramembranosa x endocondral; Estrutura histológica do osso normal vs alterado; Comprometimento das células osteoprogenitoras e osteoblastos; Possíveis alterações na remodelação óssea; Relação entre histologia e manifestações clínicas.
-Tipos de ossificação e sua relação com a DCC:
- Ossificação intramembranosa
Processo em que o osso é formado diretamente a partir do tecido mesenquimal, sem passar por um estágio cartilaginoso.
Responsável pela formação de ossos como clavículas, crânio, maxila e partes da mandíbula (exceto o côndilo).
Altamente dependente da função do gene RUNX2, essencial para a diferenciação de osteoblastos.
Na DCC, esse processo é profundamente comprometido, levando à formação de ossos finos, porosos, com suturas abertas e clavículas ausentes ou malformadas.
- Ossificação endocondral
Processo em que um molde cartilaginoso é gradualmente substituído por osso.
Presente nos ossos longos como fêmur, tíbia, e também no côndilo da mandíbula.
RUNX2 também participa da transição dos condrócitos para osteoblastos nesse processo, mas com menor impacto do que na ossificação intramembranosa.
Na DCC, pode haver atraso ou leve encurtamento de ossos longos, mas geralmente sem deformidades significativas.
-Estrutura histológica do osso na DCC:
O osso normal é predominantemente lamelar, com matriz osteoide bem organizada e rica em colágeno tipo I, além de ser eficientemente mineralizado com deposição de cristais de hidroxiapatita.
- Na DCC, a matriz é desorganizada e hipomineralizada, com predomínio de osso entrelaçado (imaturo), o que compromete a resistência mecânica do osso.
- Osteoblastos maduros são escassos ou ausentes, e as superfícies ósseas podem conter células imaturas que não completam a função de formação óssea.
- As trabéculas ósseas são finas, porosas e distribuídas de forma irregular, refletindo um processo de ossificação incompleto.
- Regiões que normalmente conteriam osso maduro podem ser substituídas por tecido conjuntivo fibroso, especialmente nas clavículas e crânio.
-Comprometimento das células osteoprogenitoras e osteoblastos
- Células osteoprogenitoras
São células indiferenciadas de origem mesenquimal com potencial para se tornarem osteoblastos. Na DCC, há uma falha na diferenciação dessas células devido à mutação no gene RUNX2, que atua como fator de transcrição necessário para essa transformação. Como resultado, o número de osteoblastos maduros é drasticamente reduzido.
- Osteoblastos
Responsáveis por produzir a matriz óssea (colágeno tipo I) e iniciar o processo de mineralização com secreção de fosfatase alcalina e osteocalcina. Na DCC, os poucos osteoblastos formados não atingem funcionalidade plena. A produção de matriz é deficiente, assim como sua mineralização, levando à persistência de tecido osteoide mal mineralizado.
-Alterações na remodelação Óssea
- A remodelação óssea é um processo dinâmico de reabsorção (por osteoclastos) e formação (por osteoblastos), essencial para manter o equilíbrio estrutural do osso.
- Na DCC, os osteoclastos funcionam normalmente, mas a formação óssea está comprometida pela deficiência osteoblástica.
- O desequilíbrio entre reabsorção e formação leva à redução da densidade óssea, irregularidade das trabéculas e falha no fechamento das suturas cranianas.
- Há também comprometimento da remodelação do osso alveolar, o que afeta diretamente a erupção dentária.
- Como o osso não é reabsorvido adequadamente, os dentes permanentes permanecem impactados, mesmo com raízes completas.
-Relação entre histologia e manifestações clínicas
- A deficiência da ossificação intramembranosa explica a presença de fontanelas abertas, suturas cranianas persistentes e ausência ou hipoplasia das clavículas.
- A fragilidade do osso entrelaçado mal mineralizado contribui para o formato anormal do crânio (em tenda), deformidades faciais e alterações posturais.
- A hipoplasia da maxila e a protrusão mandibular relativa decorrem da assimetria de desenvolvimento facial, ligada à matriz óssea mal formada.
- A falha na remodelação óssea impede a erupção normal dos dentes permanentes e favorece a retenção de dentes decíduos.
- A persistência da lâmina dentária resulta na formação de múltiplos dentes supranumerários, frequentemente impactados.
- A ausência de osso funcional em determinadas regiões pode levar à substituição por tecido fibroso, como nas articulações dos ombros, permitindo uma mobilidade anormal devido à ausência de clavículas.
3 – Alterações odontológicas e repercussões clínicas
Agenesia dentária (ausência de dentes); Retenção de dentes decíduos e inclusões dentárias; Hipoplasia do esmalte ou má-formações; Radiografias típicas: múltiplos dentes inclusos; Dificuldades clínicas no tratamento ortodôntico e cirúrgico.
Sabe-se que a displasia cleidocraniana (CCD) pode acarretar diversas consequências para os indivíduos acometidos, afetando diferentes aspectos do desenvolvimento ósseo e dentário. Entre essas manifestações, as alterações dentárias se destacam como uma das principais características clínicas, exercendo grande impacto funcional, estético e até psicossocial, uma vez que podem interferir na mastigação, na fala e na autoestima dos pacientes.
-Principais alterações dentárias
- Dentes supranumerários: Pacientes com CCD frequentemente apresentam diversos dentes supranumerários, geralmente localizados na região anterior da maxila e mandíbula. Esses dentes adicionais dificultam ou impedem a erupção dos dentes permanentes.
- Retenção prolongada dos dentes decíduos: A presença de dentes supranumerários e a falha na reabsorção radicular impedem a esfoliação normal dos decíduos, causando atraso na substituição dentária.
- Dentes inclusos ou impactados: Muitos dentes permanentes permanecem retidos no interior do osso, mesmo após extrações de decíduos ou supranumerários. Radiografias panorâmicas costumam mostrar múltiplos dentes inclusos.
- Agenesia dentária: Em alguns casos, há ausência congênita de certos dentes permanentes, o que gera espaços na arcada e necessidade de reabilitação protética.
- Hipoplasia do esmalte: Alguns dentes apresentam esmalte fino ou defeituoso, predispondo a cáries, desgaste precoce e fraturas coronárias.
-Repercussões clínicas
As alterações odontológicas da CCD provocam:
- Má oclusão dentária, frequentemente associada a discrepâncias ósseas maxilomandibulares;
- Comprometimento funcional, dificultando mastigação e fala;
- Prejuízo estético, com impacto negativo na autoestima e no convívio social;
- Desafios terapêuticos, já que a movimentação de dentes inclusos requer tração ortodôntica associada a procedimentos cirúrgicos, muitas vezes em várias etapas.
-Abordagem terapêutica
- O tratamento é complexo e exige planejamento interdisciplinar, envolvendo ortodontistas, cirurgiões bucomaxilofaciais e, em alguns casos, protesistas e implantodontistas. As principais etapas incluem:
- Extração dos dentes supranumerários e decíduos retidos em idade precoce;
- Exposição cirúrgica de dentes permanentes inclusos, seguida de tração ortodôntica para posicionamento na arcada;
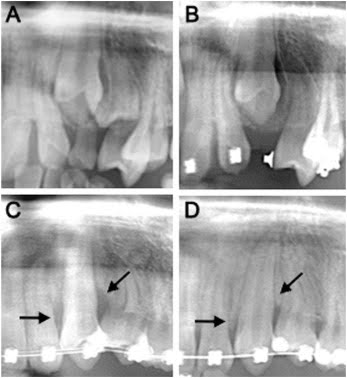
Radiografias de acompanhamento para a tração ortodôntica de dentes impactados: A, radiografia inicial do dente 23 (setembro de 2017); B, após a extração do dente decíduo retido (janeiro de 2018); C, condição do tecido periodontal durante a tração ortodôntica (dezembro de 2018); D, condições do tecido periodontal ao final do tratamento ortodôntico (setembro de 2019). As setas indicam alterações na altura de inserção óssea alveolar. (SHI, 2025)
- Restaurações ou coroas protéticas para dentes com hipoplasia de esmalte;
- Implantes ou próteses em casos de agenesia dentária.
4 – Comprometimento do sistema esquelético além do crânio: manifestações clínicas e funcionais.
Hipoplasia ou ausência total das clavículas: impacto na mobilidade escapular (ex: possibilidade de juntar os ombros na frente do tórax); Fontanelas e suturas cranianas abertas além da idade habitual; Alterações em ossos longos e pélvicos; Estatura baixa e escoliose: causas fisiopatológicas; Implicações ortopédicas e cirúrgicas.
O comprometimento esquelético além do crânio envolve alterações estruturais com repercussões clínicas e funcionais relevantes, sobretudo na biomecânica articular e na estabilidade postural. Uma das manifestações mais marcantes é a hipoplasia ou ausência completa das clavículas, permitindo mobilidade escapular aumentada, incluindo a capacidade do indivíduo de aproximar os ombros anteriormente até a linha média torácica. Embora essa característica seja compatível com preservação funcional em atividades de baixa demanda, avaliações clínicas apontam que a ausência do suporte clavicular pode provocar instabilidade dinâmica, dor por sobrecarga muscular e limitação funcional em tarefas repetitivas ou que exijam resistência prolongada. A literatura descreve impacto leve a moderado na funcionalidade dos membros superiores, frequentemente avaliado por instrumentos como a escala SPADI.
Outra manifestação recorrente é a persistência de fontanelas abertas e suturas cranianas patentes além da idade esperada de fechamento fisiológico. Esse quadro pode ser acompanhado por protuberância frontal bilateral e presença de ossos de Wormian (pequenos ossos acessórios que se formam dentro das suturas do crânio, especialmente ao longo da sutura lambdoide, resultando em centros de ossificação adicionais durante o desenvolvimento do crânio) nas suturas lambdoides e sagitais. Embora a patência prolongada dessas estruturas raramente represente risco neurológico direto, ela pode gerar alterações estéticas perceptíveis e, em alguns casos, motivar indicação cirúrgica reconstrutiva ou correção em contexto de fratura craniana.

Imagem 4: Tomografia computadorizada mostra presença de ossos de Wormian. Fontanela anterior e sutura metópica patentes em um paciente de 23 anos.
As alterações nos ossos longos e na pelve representam outro eixo crítico de comprometimento. São descritas anomalias como encurtamento femoral, coxa vara, epífises achatadas e hipoplasia das asas ilíacas, frequentemente associadas a ossificação púbica tardia. Tais alterações afetam o eixo de sustentação corporal, gerando assimetria pélvica, instabilidade biomecânica e distúrbios de marcha. Indivíduos com essas alterações podem apresentar claudicação funcional, dor lombopélvica crônica e sobrecarga compensatória de musculaturas estabilizadoras. O manejo inclui desde fisioterapia até osteotomias femorais ou reconstruções acetabulares nos casos de impacto funcional significativo.

Imagem 5: Radiografia anteroposterior da pelve mostrando encurtamento do colo femoral em ambos os lados (seta preta), deformidade da cabeça femoral em “chapéu de chef” (seta vermelha), alargamento da articulação sacroilíaca (seta azul) (A), e radiografia da pelve evidenciando coxa vara no quadril direito e coxa valga no quadril esquerdo (B).
A baixa estatura, comum nesses indivíduos, resulta de falhas na ossificação intramembranosa e, em menor grau, endocondral, afetando o crescimento longitudinal dos ossos proporcionalmente. Embora não seja, por si só, uma limitação funcional, a estatura reduzida pode intensificar desequilíbrios estruturais e contribuir para alterações posturais, como a escoliose. Esta, por sua vez, é frequentemente identificada em fases precoces do desenvolvimento e pode progredir durante a puberdade, exigindo monitoramento radiográfico regular. Curvaturas torácicas ou toracolombares, quando acentuadas, podem comprometer o alinhamento axial e, em casos graves, levar a dor crônica ou alterações da mecânica ventilatória. A conduta terapêutica varia conforme a gravidade, podendo envolver ortetização ou intervenção cirúrgica com artrodese vertebral.
As implicações ortopédicas do comprometimento esquelético extracefálico são amplas e exigem abordagem multidisciplinar. A reconstrução clavicular pode ser considerada em casos de instabilidade funcional severa, enquanto deformidades pélvicas e femorais podem requerer correções ósseas cirúrgicas. Já as alterações vertebrais demandam uma avaliação integrada entre ortopedia, fisioterapia e, em alguns casos, cirurgia da coluna. A tomada de decisão deve considerar fatores como idade óssea, grau de deformidade, funcionalidade atual e potencial de progressão.
5 – Referências
- FARROW, Emily G. et al. Craniofacial and orthopedic management of cleidocranial dysplasia. Journal of Craniofacial Surgery, [s. l.], v. 29, n. 8, p. 1925–1930, 2018. Disponível em: https://ern-ithaca.eu/wp-content/uploads/2020/12/Farrow_CCD_general_JCraniofacSurg2018.pdf. Acesso em: 20 jul. 2025.
- GÓMEZ VIEIRA, Rafael; LIMA, Soraia; SILVA, Maria Eduarda. Functional impact of clavicular hypoplasia in shoulder mobility: case report and literature review. International Journal of Medical Science and Clinical Inventions, [s. l.], v. 4, n. 10, p. 3196–3198, 2017. Disponível em: https://valleyinternational.net/index.php/ijmsci/article/view/971. Acesso em: 20 jul. 2025.
- JIRAPINYO, Peerasak et al. Orthopedic and radiographic assessment of long bone and pelvic abnormalities in cleidocranial dysplasia. Journal of Craniofacial Surgery, [s. l.], v. 31, n. 8, p. 2321–2325, 2020. Disponível em: https://ern-ithaca.eu/wp-content/uploads/2020/12/Jirapinyo_CCD_bone_JCraniofacSurg2020.pdf. Acesso em: 20 jul. 2025.
- KOMORI, T. Regulation of osteoblast differentiation by transcription factors. Journal of Cellular Biochemistry, [s. l.], v. 106, n. 6, p. 927–932, 2010. DOI: https://doi.org/10.1002/jcb.22052.
- MOORE, Keith L.; PERSAUD, T. V. N.; TORCHIA, M. G. Embriologia clínica. 10. ed. Rio de Janeiro: Elsevier, 2020.
- ONUR, Ozlem Ecevit et al. Scoliosis and spinal deformities in skeletal dysplasias: case series and review. Clinical Case Reports, [s. l.], v. 8, n. 12, p. 2831–2835, 2020. Disponível em: https://onlinelibrary.wiley.com/doi/full/10.1002/ccr3.6440. Acesso em: 20 jul. 2025.
- ORTHOBULLETS. Cleidocranial dysostosis (CCD). Orthobullets: Pediatrics, [s. l.], 2023. Disponível em: https://www.orthobullets.com/pediatrics/4100/cleidocranial-dysplasia-dysostosis. Acesso em: 20 jul. 2025.
- OTTO, F. et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell, [s. l.], v. 89, n. 5, p. 765–771, 1997. DOI: https://doi.org/10.1016/S0092-8674(00)80259-7.
- ROBBINS, Vinay Kumar; COTRAN, Ramzi S. Patologia: bases patológicas das doenças. 10. ed. Rio de Janeiro: Elsevier, 2021.
- SADLER, T. W. Langman: embriologia médica. 14. ed. Rio de Janeiro: Elsevier, 2021.
- SANJAY, A. et al. Genetic disorders of bone: RUNX2 and cleidocranial dysplasia. Frontiers in Endocrinology, [s. l.], v. 11, p. 619346, 2020. DOI: https://doi.org/10.3389/fendo.2020.619346.
- SHI, Y.; REN, J.; WANG, K.; LIU, L.; WANG, H.; YOU, M. Exploring the complexities of cleidocranial dysplasia: dental anomalies and treatment interventions. American Journal of Orthodontics and Dentofacial Orthopedics, [s. l.], v. 167, n. 3, p. 319-330.e2, mar. 2025. DOI: https://doi.org/10.1016/j.ajodo.2024.10.014. Disponível em: https://pubmed.ncbi.nlm.nih.gov/39674929/. Acesso em: 26 jul. 2025.