
Tema: Síndrome de Treacher Collins
Extraordinário é um filme tocante que narra a história de Auggie Pullman, um menino que nasceu com uma síndrome genética rara, resultando em uma deformidade facial. Após passar por inúmeras cirurgias e anos de ensino domiciliar, Auggie se prepara para enfrentar um grande desafio: ingressar em uma escola regular. A trama aborda temas de aceitação, empatia e superação, mostrando a jornada de Auggie e como ele e sua família lidam com as dificuldades sociais e emocionais decorrentes de sua condição.
Nosso novo artigo explora a condição genética retratada no filme, discutindo as causas, sintomas e tratamentos disponíveis. Além disso, fazemos uma análise crítica sobre a representação de tais condições na mídia e como isso pode impactar a percepção pública. Convidamos você a ler o conteúdo completo para entender melhor a ciência por trás de “Extraordinário” e a realidade das pessoas que vivem com essa condição.
Definição e história
A síndrome de Treacher Collins (disostose mandibulofacial) é um distúrbio autossômico dominante caracterizado por hipoplasia malar (subdesenvolvimento dos ossos zigomáticos da face) com fissuras palpebrais com inclinação para baixo, defeitos das pálpebras inferiores, orelhas externas deformadas e, às vezes, defeitos das orelhas média e interna.
O gene 1 da síndrome de Treacher Collins-Franceschetti (TCOF1) é responsável pela produção de uma proteína chamada treacle. Essa proteína está envolvida na biogênese do RNA ribossômico que contribui para o desenvolvimento dos ossos e das cartilagens da face. A mutação no gene TCOF1 está associada à síndrome de Treacher Collins.
Embora Thomson tenha sido o primeiro a referenciar esta síndrome em 1846, foi E. Treacher Collins quem descreveu seus componentes essenciais em 1900. Franceschetti e Klein, em 1949, realizaram intensivos estudos da síndrome. Além de Síndrome de Treacher Collins, epônimo preferido pela literatura inglesa, é referida também como disostose mandibulofacial, Síndrome de Berry e Síndrome de Franceschetti-Zwahlen-Klein.
Etiologia e Genética
Os ribossomos sintetizam todas as proteínas e, portanto, são críticos para o crescimento e proliferação celular. A biogênese do ribossomo, ou o processo de produção de ribossomos, é um dos processos que mais consomem energia dentro de uma célula, e interrupções na biogênese do ribossomo podem levar a distúrbios congênitos denominados ribossomopatias. Curiosamente, as ribossomopatias individuais são caracterizadas por fenótipos específicos do tecido, o que é surpreendente, dada a importância universal dos ribossomos. A síndrome de Treacher Collins (TCS), por exemplo, é uma ribossomopatia caracterizada por anomalias dos ossos faciais, palato, olhos e ouvidos.
A síndrome de Treacher Collins geralmente é causada devido a mutações nos genes TCOF1, POLR1B, POLR1C ou POLR1D. Estas mutações normalmente acontecem ao acaso, mas podem ser passadas de pais para filhos, especialmente quando existem casos da síndrome na família. Geralmente, caso um dos pais tenha a síndrome, a probabilidade dos filhos desenvolverem a doença é de 50%.
TCOF1 (Treacle): O gene TCOF1 está envolvido na síntese de uma proteína chamada treacle. Mutações nesse gene causam a síndrome de Treacher Collins, uma condição rara que afeta o desenvolvimento craniofacial. Os sintomas incluem anomalias nos ossos da face, palato, olhos e orelhas.
POLR1C e POLR1D: Esses genes codificam subunidades das RNA polimerases I e III, que são essenciais para a transcrição dos RNAs ribossômicos (rRNAs). Perturbações na biogênese dos ribossomos podem levar a defeitos específicos em tecidos, conhecidos como ribossomopatias. No caso da síndrome de Treacher Collins, mutações em POLR1C e POLR1D resultam em defeitos craniofaciais, como hipoplasia da cartilagem e anomalias no esqueleto craniano. Além disso, a deficiência de migração das células da crista neural, que são progenitoras do esqueleto craniofacial, também está associada a essas mutações.
Características Clínicas
A Síndrome de Treacher Collins é uma condição genética rara que afeta o desenvolvimento dos ossos e tecidos do rosto. As características clínicas desta síndrome são variadas e podem incluir deformidades faciais, como a ausência ou subdesenvolvimento dos ossos das bochechas, mandíbula e queixo. Outras manifestações comuns são as fendas palpebrais inclinadas para baixo, colobomas nas pálpebras inferiores, e malformações das orelhas externas, que podem levar a perda auditiva. Além disso, alguns pacientes podem apresentar problemas respiratórios devido a vias aéreas estreitas. A gravidade dos sintomas pode variar amplamente entre os indivíduos, mas essas características clínicas são fundamentais para o diagnóstico e manejo da condição.
Em suma, podemos elencar as principais sintomatologias clínicas:
- Olhos caídos, com as pálpebras inclinadas para baixo;
- Queixo pequeno;
- Alterações nas orelhas, como deformidades, orelhas pequenas ou ausentes;
- Ausência de cílios;
- Pálpebras inferiores pouco desenvolvidas;
- Crescimento de cabelo próximo das “maçãs” do rosto;
- Dificuldade para ouvir;
- “Maçãs” do rosto pequenas;
- Boca seca, devido à menor produção de saliva;
- Fenda no lábio ou céu da boca em alguns casos;
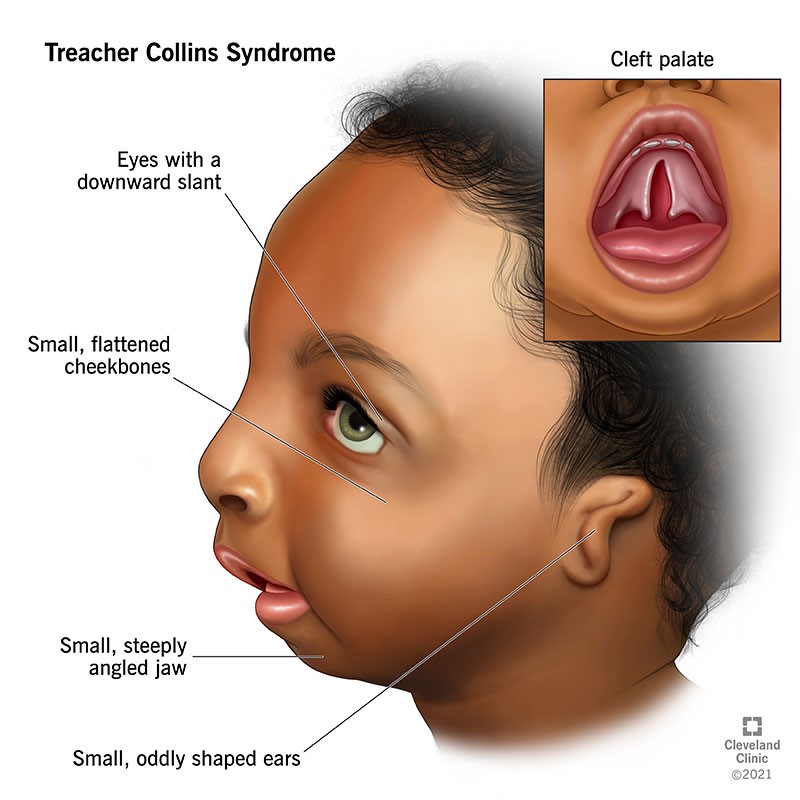

Devido às alterações nos ossos da face, pode existir dificuldade para se alimentar ou respirar adequadamente. Além disso, dependendo das alterações físicas, o paciente também pode desenvolver problemas como depressão ou ansiedade.
Embora a dificuldade para ouvir seja comum em pessoas com a síndrome, geralmente não causa atraso de desenvolvimento em crianças ou afete a capacidade de raciocínio do paciente.
Diagnóstico
Este distúrbio hereditário é caracterizado por inclinação antimongolóide das fissuras palpebrais, coloboma da pálpebra inferior, micrognatia e hipoplasia das arcadas zigomáticas e micrognatia e hipoplasia das arcadas zigomáticas e microtia. Podem estar incluídos no quadro clínico , a inclinação descendente das fendas palpebrais, ptose palpebral, ausência de velum (raramente com atresia das coanas), hipoplasia da maxila com fenda do palato secundário ou palato arqueado. As fendas labiais são raras.
Ao nascimento, essas crianças devem ser examinadas pelo otorrinolaringologista e pelo
fonoaudiólogo a fim de se identificar a extensão da perda auditiva e o grau de comprometimento
das vias aéreas. Nos indivíduos brandamente afetados o diagnóstico necessita da comprovação genética. Esse diagnóstico pode ser feito no pré-natal através de biópsia do vilo corial entre 10º
e 13º semanas de ou amniocentese entre 16º e 18º semanas de gestação para realização de estudo
genético. A fetoscopia e a ultrassonografia são métodos auxiliares no diagnóstico.
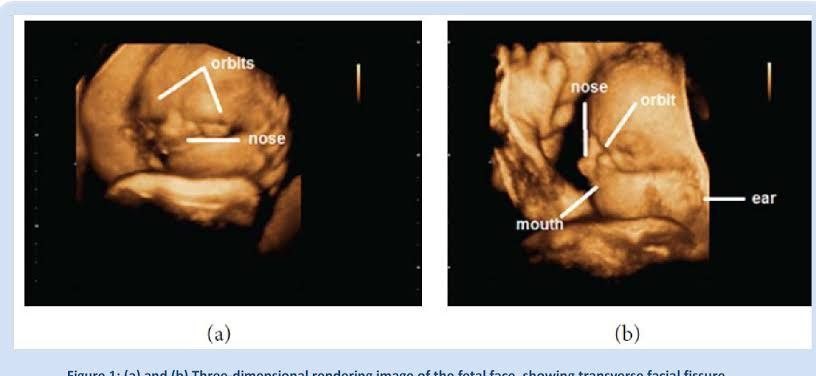
(a) e (b) renderização tridimensional de imagem de face fetal, mostrando fissura facial transversa, microftalmia e orelhas baixas
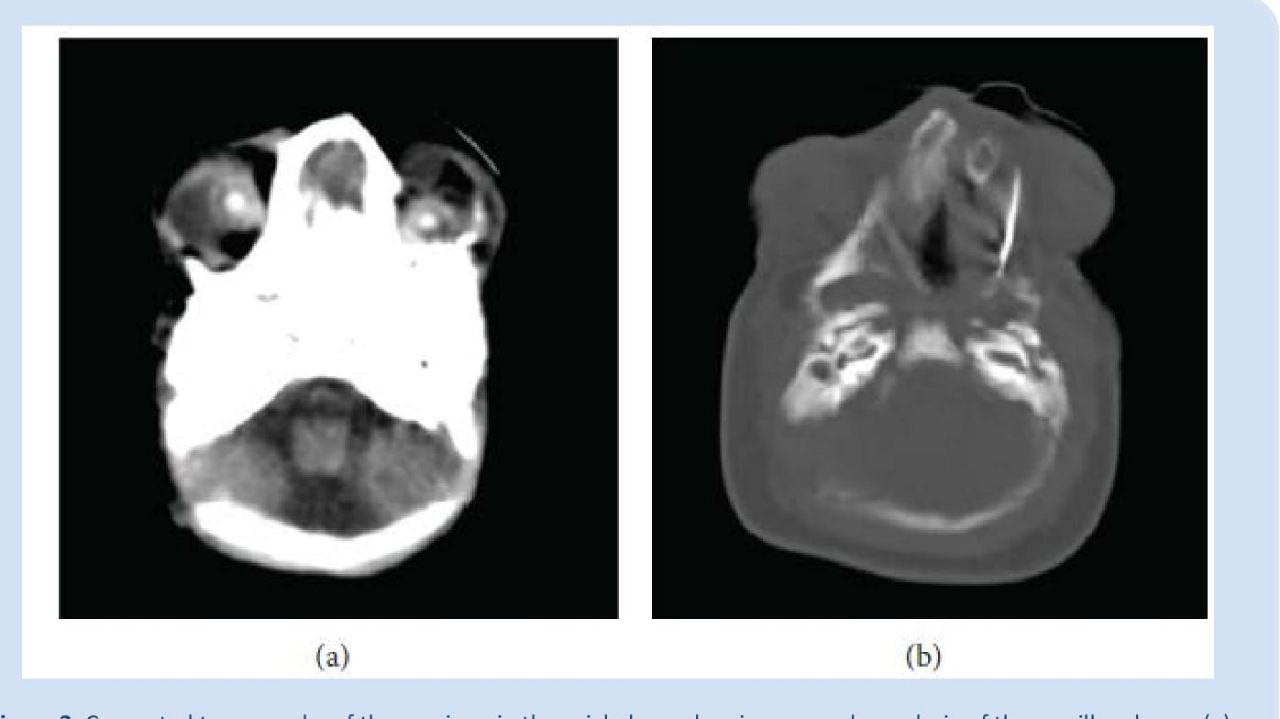
Tomografia computadorizada do crânio, em plano Axial, mostrando severa hipoplasia dos ossos maxilares (a), os quais estão dismórficos e cobertos com pele (b)
Tratamento e Manejo
Essa síndrome não tem cura, o tratamento eficiente é através de uma equipe multidisciplinar, que inclui: Cirurgião craniofacial; Otorrinolaringologista; Oftalmologista; Fonoaudiólogo; Odontólogo; Terapeuta ocupacional; Psicólogo e Psiquiatra.
Através dos métodos cirúrgicos pode-se corrigir algumas deformidades dos ossos da face, para melhorar a função e a aparência; Pode ser necessário algumas cirurgias de reconstrução auricular e de vias aéreas, assim como a ortognática para corrigir problemas da mordida e alinhar a mandíbula.
Em alguns casos pode ser necessário também o uso de aparelhos auditivos ou até implantes cocleares, juntamente com terapias de fonoaudiologia e terapia ocupacional. Essas ações ajudam o paciente a melhorar a audição, a fala e a comunicação e consequentemente o desenvolvimento motor e funcional do portador da síndrome.
Cuidados com o Oftalmologista são importantes para tratar problemas de visão e evitar problemas futuros. Já os cuidados dentários são fundamentais, pois pela alteração anatômica da face, é necessário tratamento para para alinhar os dentes e corrigir problemas de mordida, assim como manter a saúde bucal.
O suporte Psicológico e Psiquiátrico ajuda a lidar com os aspectos emocionais e sociais da condição. Existem Grupos de Apoio para pacientes e famílias compartilharem experiências e receberem suporte.
O manejo da Síndrome de Treacher Collins é um processo contínuo e personalizado, adaptado às necessidades individuais de cada paciente com monitoramento e avaliação dos profissionais da equipe multidisciplinar, para acompanhar o desenvolvimento e a eficácia dos tratamentos e conforme necessário, à medida que a criança cresce e se desenvolve.
Prognóstico e qualidade de vida
A síndrome de Treacher Collins não é uma doença progressiva e, geralmente, as crianças afetadas alcançam normal desenvolvimento e inteligência. O prognóstico das formas leves e moderadas da doença é favorável com a implementação de tratamento adequado.
A presença de distúrbios respiratórios, relacionados com condição, podem impactar a qualidade de vida ao promoverem alteração no sono, gerando um quadro de morbidade que afeta o sistema nervoso central das pessoas nessa situação
No que se refere a estética, pode-se dizer que além da alteração funcional, ocorrem problemas sociais ao indivíduo com a síndrome. Tal panorama é fruto dos preconceitos que permeiam a STC resultando em ofensas no ambiente escolar, principalmente, mas também em outros locais públicos. Consequentemente, indivíduos com Treacher Collins, apresentam elevadas taxas de patologias psicológicas, como ansiedade, depressão e insegurança.
Pesquisa atual e Avanços
Atualmente, os avanços alcançados nas pesquisas sobre a Síndrome de Treacher Collins (STC) envolvem diferentes áreas-chave devido ao progresso na compreensão dos mecanismos genéticos, diagnósticos e possíveis tratamentos. Entre os principais, cabe citar:
- Terapias Gênicas e Edição Genética
Presentemente já se sabe que mutações nos genes TCOF1, POLR1C e POLR1D estão relacionadas com o desenvolvimento da STC. Pesquisas recentes estão sendo responsáveis por identificar novas variantes genéticas e possíveis mutações adicionais que podem contribuir para o surgimento dessa patologia. Com esse intuito, são utilizadas principalmente técnicas de edição de determinados genes em laboratório, além do uso de modelos animais, como roedores e até algumas espécies de peixes, para realização de testes mais conclusivos.
- Avanços no diagnóstico
É certo que o diagnóstico e o planejamento do tratamento precoces da síndrome de Treacher Collins são essenciais para garantir não somente a sobrevivência do paciente, mas também proporcionar qualidade de vida e dignidade enquanto isso. Atualmente, isso já é possível pela existência de um vasto conhecimento dos profissionais da saúde que estão inseridos no mercado de trabalho, mas também pelos métodos de diagnósticos eficientes que são adotados baseados na idade do paciente, o que permite traçar um plano de tratamento objetivo, focado nas demandas e atento aos riscos que cada caso pode apresentar. Além disso, o entendimento atual da necessidade de uma equipe extremamente competente, especializada e multidisciplinar (contando com pediatras, geneticistas, otorrinolaringologistas, ortodontistas, fonoaudiólogos, psicólogos e uma grande diversidade de cirurgiões) garante que importantes avanços sejam feitos em relação a essa condição complexa e intrigante.
- Terapias de reabilitação
Com o passar das décadas e o desenvolvimento de tecnologias inovadoras, novas formas de tratamento e recuperação tem influenciado positivamente na qualidade de vida de pacientes afetados com essa condição sindrômica em todo o mundo, entre alguns exemplos desses avanços é possível destacar a modelagem e impressão 3D de modelos que visam promover a reconstrução de estruturas prejudicadas do rosto (ossos, cartilagens e outros tipos de tecidos), garantindo uma melhoria tanto estética quanto funcional para os pacientes atendidos.